
Las cada vez más numerosas opciones de tratamiento innovadoras en el área de la oncología se deben, en gran parte, a las empresas biotecnológicas, cuya cartera de I+D representa un 70 por ciento. Pero estas empresas se enfrentan a grandes desafíos, pues no solo tienen la tarea de desarrollar terapias innovadoras, sino también asegurar fondos de inversión para mantener sus esfuerzos de investigación.
Ante ello, IQVIA explora las últimas tendencias en cáncer y orienta al sector a la hora de optimizar los ensayos clínicos a través del documento ‘Navegando por el complejo panorama oncológico’, basado en el informe ‘Tendencias globales en oncología 2023’ publicado en el mes de mayo de este año.
De este modo, la consultora analiza el diseño y la estrategia de los ensayos oncológicos en fase inicial y el diseño para los ensayos de plataforma que pueden ampliar el acceso a nuevas opciones de tratamiento. Asimismo, examina los esfuerzos realizados para mejorar la representación racial y étnica en los ensayos y realiza una serie de recomendaciones para incorporar la planificación de la diversidad en los protocolos de los ensayos clínicos.
Aumento de ensayos clínicos
Los ensayos clínicos de oncología han observado un aumento en los niveles de actividad a pesar de la interrupción que se produjo durante la pandemia. En 2022, se lanzaron aproximadamente 2.300 nuevos ensayos, lo que supone un aumento del 30 por ciento desde 2017. Según IQVIA, para 2022, el crecimiento se había estabilizado, pero el número total de ensayos iniciados ese año estableció nuevos récords.
Además, la línea de desarrollo ha experimentado una transformación, atribuida al aumento de bioterapéuticos de próxima generación. “Los anticuerpos inmunoconjugados (ADC) y los anticuerpos biespecíficos han ido ganando interés, especialmente en el tratamiento de tumores sólidos”, apunta el documento. Desde 2020, los inmunooncológicos han mantenido una presencia constante en los enfoques de tratamiento del cáncer y las terapias celulares, CAR-T, terapias de ARN, vacunas, edición genética y terapia génica han ido ganando terreno.
La contribución de las empresas biofarmacéuticas emergentes es evidente no solo en el desarrollo de medicamentos, sino también en las solicitudes de aprobación regulatoria y los lanzamientos al mercado. De hecho, en 2022, estas empresas fueron responsables de introducir 7 de las 10 sustancias lanzadas.
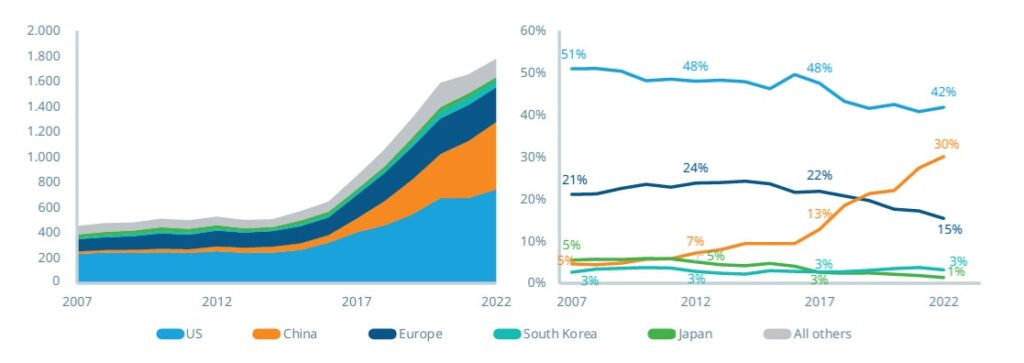
Pero, geográficamente, mientras que algunos países han experimentado un aumento de las contribuciones, otros han quedado más rezagados. China cuenta con el 30 por ciento de la cartera mundial de medicamentos oncológicos en desarrollo. Por su parte, empresas con sede en Estados Unidos y Europa han disminuido su participación, incluso “cuando el número absoluto de moléculas en desarrollo ha aumentado”.
Incertidumbre en el desarrollo clínico
A pesar del avance por parte de las empresas biotecnológicas a la hora de desarrollar nuevas terapias, se enfrentan a numerosos desafíos por el camino. El documento menciona algunas incertidumbres del desarrollo clínico de oncología, como la baja tendencia de las tasas de éxito, la complejidad de los ensayos, los retos de financiación y los cambios regulatorios.
Tal y como señala el texto, de 2016 a 2022, las tasas de éxito en el desarrollo clínico de oncología han mostrado una tendencia a la baja en las categorías tanto raras como no raras, sólidas y hemo/oncológicas. Esto puede deberse a que “las empresas asuman un mayor riesgo científico en sus programas clínicos, impulsadas por un mayor apetito por avances tanto desde la perspectiva regulatoria como de los resultados de los pacientes”. Sin embargo, añaden, este mayor riesgo científico también conduce a un mayor riesgo de fracaso, especialmente en la Fase II y más allá.
Por otro lado, los ensayos en oncología plantean un desafío por su complejidad, que muchas veces supera a otras áreas terapéuticas. Esta complejidad se refleja en la duración del estudio, el recuento de participantes, el número de sitios y países, los criterios de elegibilidad y los criterios de valoración. Según IQVIA, “si bien es posible un rápido desarrollo en oncología, la mayoría de las moléculas tardan entre seis y diez años” en lanzarse al mercado. En este sentido, un objetivo ambicioso es reducir los plazos para lanzar más moléculas dentro de los diez años posteriores a la solicitud de patente original.
En cuanto al reto de la financiación, en 2020 y 2021 se experimentó un aumento excepcional de los niveles de financiación (135.000 millones de dólares y 118.000 millones de dólares, respectivamente) para el sector biofarmacéutico, incluido el área de oncología, pero la caída a niveles más normales en 2022 y 2023, que vuelven a estar en línea con la tendencia a largo plazo, provoca incertidumbre para el sector.
Finalmente, los recientes cambios normativos están afectando al sector farmacéutico. En Estados Unidos, la Ley de Reducción de la Inflación ha generado preocupación sobre sus efectos en la industria; además, la nueva legislación exige que se presenten planes de acción sobre diversidad a la FDA antes de que comiencen los ensayos fundamentales. Y, en Europa, la nueva Estrategia Farmacéutica está despertando interés con sus importantes iniciativas y posibles reformas, aunque aún está en proceso de desarrollo legislativo.
Cómo optimizar los ensayos
Gerhard du Toit, Global Oncology Head IQVIA Biotech y uno de los autores del documento, apunta que se ha prestado mayor atención a diseños de ensayos novedosos y complejos y, en los últimos años, más de una cuarta parte de los ensayos de calidad respaldados por la industria han incorporado dichos diseños, en particular protocolos maestros.

“La industria apoya estas innovaciones en el diseño y ejecución de ensayos clínicos porque pueden ayudar a acelerar el desarrollo de fármacos”, expone el experto. En este sentido, Du Toit apunta que los protocolos maestros son fundamentales para ejecutar y optimizar programas clínicos complejos. “Ayudan a minimizar los riesgos de desarrollo al facilitar decisiones tempranas, reducir los tiempos de ciclo y los costes para una activación más rápida del estudio y distribuir los gastos de infraestructura entre múltiples ramas o subprotocolos”, explica.
Los marcos de diseño de los protocolos maestros se pueden adaptar para alinearse con los objetivos de un programa, optimizando el desarrollo clínico. Así, es necesario conocer la diferencia entre las pruebas de canasta, paraguas y plataforma:
- Ensayos de canasta: implican un único fármaco dirigido a múltiples áreas. Pueden variar desde estudios básicos de aumento de dosis sólidas hasta complejos estudios de canasta de Fase I en múltiples neoplasias malignas de células, que utilizan expansiones de subestudios, cohortes de eficiencia y datos del mundo real para atender a esta población. Este diseño también permite la incorporación de diferentes elementos dentro de un ensayo específico.
- Ensayos en paraguas: prueban múltiples agentes contra el mismo tipo de tumor, lo que permite una atención estándar compartida para los pacientes de control. Estos pueden ser utilizados por una sola compañía farmacéutica o de biotecnología para probar un grupo de agentes o un objetivo único y definido, ya sea a nivel de indicación o con medicina de precisión.
- Ensayos de plataforma: se basan en los beneficios de los estudios generales y, a menudo, incluyen elementos de diseño adaptativo. Esto se logra mediante la colaboración entre empresas de biotecnología, asociaciones de pacientes y organizaciones sin fines de lucro para desarrollar e invertir en terapias desatendidas compartiendo costes y riesgos.
Por otro lado, contar con diseños de ensayos flexibles y adaptables en estudios oncológicos de fase temprana “permite la revisión de datos emergentes de seguridad”. Pero existen algunos retos y Du Toit insta a tener en cuenta tres criterios principales: la selección del país o lugar donde se realizará el ensayo, la puesta en marcha y la excelencia de los datos y la toma de decisiones.
Del mismo modo, cada vez está cobrando más importancia la diversidad y la inclusión en los ensayos clínicos, especialmente en Estados Unidos, convirtiéndose en el foco de reguladores y partes interesadas. “Creemos que todos los involucrados en los ensayos clínicos tienen la responsabilidad de aumentar el acceso para las poblaciones históricamente desatendidas. Esto incluye comprender y abordar las barreras, examinar los criterios de inclusión/exclusión y evaluar la selección del sitio”, afirma Gerhard du Toit.
Finalmente, el experto apunta a la adopción de un enfoque de ciclo de vida para lograr esa diversidad en los ensayos, que comienza con “una cuidadosa consideración del diseño del ensayo y la selección del sitio, continúa a través de la activación del sitio y respalda las expectativas de comunicación asociadas con el reclutamiento de pacientes, lo que culmina en la retención de datos”.







